NGS基因检测,癌症肿瘤NGS检测:MRD、TMB、MSI、HRD
一、MRD检测
MRD具有多种表述:微小残留病灶(minimal residual disease)、可测量残留病灶(measurable residual disease)和分子残留病灶(molecular residual disease)。其概念由血液肿瘤逐步延伸至实体肿瘤, 主要指经过治疗后, 传统影像学或实验室方法不能发现, 但通过液体活检发现的癌来源分子异常, 是体现在治疗后体内存在残留肿瘤细胞的生物标志物。由于残留的癌细胞数量较少, 无法通过传统方法检测到。因此, MRD检测对于检测技术的敏感性有极高的要求。随着分子生物学技术的发展, 特别是NGS技术的深入应用, 对MRD的检测已应用到大量临床研究中。
在实体肿瘤中, MRD与患者术后生存复发之间相关性证据已在乳腺癌、消化道肿瘤、肺癌等中得到印证。基于ctDNA的MRD检测已成为临床越发关注的一个重点。很多研究表明MRD能够早于传统影像学发现肿瘤复发, 是一个优良的预后指标, 也有越来越多的证据表明MRD有望成为一个预测指标。
目前, 常用的MRD检测技术包括数字聚合酶链式反应(digital polymerase chain reaction, dPCR)、NGS等。其中, NGS作为新兴的MRD检测方法, 对于预后有更准确的评估, 已受到国内外专家认可并推荐。
基于NGS检测MRD检测的基本策略, 包括肿瘤先验分析(tumor-informed assays, 个体化定制或NGS panel)和肿瘤未知分析(tumor-agnostic assays, NGS panel和多组学技术), 两种策略目前均处在探索阶段, 需要前瞻性研究确定其敏感性、特异性和临床预测及应用价值。
虽然2021年《非小细胞肺癌分子残留病灶专家共识》已提出的标准:基于NGS的突变检测技术, 所选用的多基因panel中必须覆盖患者Ⅰ /Ⅱ 类基因变异, 基本技术标准是可稳定检测出丰度≥ 0.02%的ctDNA。
但因目前各家检测机构其技术路线、检测性能、生信分析、MRD阳性标准尚不能统一, 业界也尚未形成标准, 故目前尚无法对MRD检测报告形成统一的解读模式。
二、TMB检测
肿瘤突变负荷(tumor mutational burden, TMB)即肿瘤基因组编码区包含的非同义突变的数量或密度(突变数/Mb), 是肿瘤新抗原负荷的替代指标, 过去以全外显子组测序(whole-exome sequencing, WES)作为评估的金标准。目前基于NGS大panel检测的TMB已可达到与WES的高度相关并在大量免疫治疗临床研究中证实其对疗效的预测价值。目前, 业界对组织TMB检测有几点共识:
(1)编码区覆盖大于1 Mb(大概相当于300个以上基因的全外显子区域), 最低有效测序深度应≥ 500× 。建议进行TMB检测的靶向测序panel尽可能涵盖患者更多的其他分子遗传信息, 包括可指导靶向治疗的驱动基因突变、免疫治疗可能相关的正向预测因子及负向预测因子;
(2)基于靶向测序panel的TMB检测应以WES检测为金标准, 纳入影响蛋白质编码的体细胞突变, 应保证检出突变频率≥ 5%的体细胞突变, 以保证TMB检测值的准确性和稳定性。Panel检测区域可影响TMB值, 应通过至少1 000例WES数据予以校正。同时建议使用对照样本过滤胚系变异;
(3)同一份样本采用不同检测平台、不同大小panel、不同的生物信息学算法, 都可能导致TMB绝对数值的差异。因此, 在缺少桥连试验的前提下, 直接采用某一家公司/产品的肿瘤突变负荷高(TMB high, TMB-H)界值(cut-off)来定义其他检测产品的TMB-H是不合适的;
(4)TMB值在不同癌种中存在显著差异, 应依据免疫检查点抑制剂(immune check-point inhibitors, ICI)临床疗效确定阈值, 才能最大可能筛选出ICI治疗的潜在获益人群。
由于TMB检测过程中受到诸多因素影响, 包括生物学特性(样本类型, 肿瘤类型等), 分析前因素(样本质量、数量等), 测序因素(DNA捕获区域, panel大小, 富集方法, 测序深度, 测序平台), 生信分析(突变类型, 胚系变异过滤等)及阈值设定等。
因此,TMB检测标准化面临诸多挑战:产品设计方面, TMB cut-off值的设立及验证?产品预期用途是单药还是联合用药?预测疗效还是预后?标准化方面也有很多困境:不用肿瘤的阈值的标准化; 不同算法的标准化; 性能指标验证方法的可操作性等。
因此, 国际上已经建立了TMB基因检测产品的标准化评估体系, 如美国的FoCR(Friends of Cancer Research)项目和欧洲的QuiP(Quality in Pathology)项目。FoCR使用11个实验室检测TMB平台, 不同的TMB评估参数, 为临床研究输出TMB评估推荐规范。
FoCR已经发布了第一期研究成果, 通过使用来自32种癌症类型的癌症基因组图谱(The Cancer Genome Atlas, TCGA)的公开数据, 遵循各个检测TMB平台的独特的生物信息学规则算法, 计算出各平台NGS大panel的TMB。发现基于目标区域捕获的NGS大panel与WES具有一定的相关性, 同时对于NGS大panel TMB检测的分析验证也给出了建议。
同样, QuiP项目是基于6个NGS大panel, 15个实验中心, 对来自3种肿瘤类型(肺腺癌、头颈部鳞状癌和结肠腺癌)总共20个福尔马林固定石蜡包埋(formalin-fixed paraffin-embedded, FFPE)组织样本进行检测分析。结果显示:16个检测样品在各个实验室间和各个NGS大panel间的TMB变异较低。
对比不同实验中心检测TMB的结果, 其中87.7%的检测结果Spearman相关系数大于0.6。NGS大panel检出的TMB和WES的TMB一致率达74.9%。NGS大panel检测TMB主要影响因素包括背景噪音、DNA投入量、测序深度、基因组覆盖区域和检测下限。
QuiP提供了TMB检测的真实世界数据, 研究结果提示参与项目的各个NGS大panel及实验平台可用于检测评估TMB, 但需要仔细控制评估TMB的重要参数。FoCR项目与QuiP项目正在努力探索建立TMB检测的规范框架和蓝图。
除了欧美发起的TMB一致性评价项目外, 由中国国家癌症中心中国医学科学院肿瘤医院、中国食品药品检定研究院以及中国抗癌协会肿瘤病理专业委员会联合发起的中国TMB标准化项目(China T
MB Harmonization Project)也于2020年10月18日启动, 项目旨在推动中国TMB检测一致性评估与标准化。该项目拟通过对TMB在肿瘤免疫治疗临床应用中所涉及的相关问题进行全面考察和分析, 完成对不同基因检测产品的分析和评估工作; 建立TMB计算方法在不同类型检测方法与WES之间的转换关系; 应用临床试验样本, 结合随访数据, 划定不同癌种免疫治疗的TMB最佳获益基线, 建立TMB临床检测标准及评估体系。
项目在方案流程上一定程度地参考了FoCR与QuiP积累的经验, 同时根据中国临床检测的实际情况进行了大量的优化调整。项目总共分为2个阶段, 第一阶段是中国TMB检测现状调研与能力评估, 第二阶段是各癌种TMB临床获益评估与基线建立。
同时还应注意, 对TMB的准确评估(无论基于组织或血浆cfDNA样本)建立在样本满足一定肿瘤占比的基础上, 肿瘤占比过低将导致TMB的严重低估。因此, 临床医生在拿到1份NGS报告、看到TMB数值时, 应综合考虑以下因素:选择的panel(有无准确测算TMB的能力)、检出的肿瘤突变谱及其突变丰度(肿瘤占比是否足以评估TMB、突变谱特征与TMB高/低的组合是否合理)、TMB绝对值以及该数值在已检测的肿瘤样本中的相对排序等, 综合评估TMB水平及其可信度。
三、MSI检测
微卫星不稳定性(microsatellite instability, MSI)/错配修复(mismatch repair, MMR)检测对于多种实体瘤患者具有重要临床意义。过去以PCR毛细管电泳法作为MSI检测的金标准, 基于美国国家癌症研究所(National Cancer Institute, NCI)推荐的5~7个经典微卫星位点, 对比肿瘤细胞与正常细胞的检测结果, 以确定肿瘤细胞的MSI状态。
近年来, NGS panel开始用于MSI检测, 使用计算工具同时研究基因组上的大量微卫星序列成为可能。且多数NGS-MSI算法采用正常人长度分布模型, 无需正常组织作对照。1个NGS panel需要整合一定数量的有效微卫星位点、构建相应的MSI算法并经过临床样本验证, 才能准确报告MSI状态。
对MSI的准确评估同样也建立在一定肿瘤占比的基础上。对于MSI检测结果, 应综合样本肿瘤占比(可参考肿瘤突变丰度)、TMB水平[微卫星高度不稳定性(MSI-high, MSI-H)的肿瘤往往TMB-H]、肿瘤突变谱特征(MSI-H可由MMR基因失活性突变导致, 且基因突变谱往往以Indel为主)等信息综合分析, 判断其可信度。
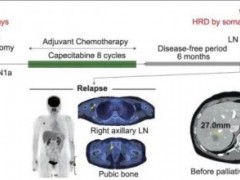
四、HRD检测
同源重组修复缺陷(HRD)通常指细胞水平上的同源重组修复(homologous recombination repair, HRR)功能障碍状态, HRD可由HRR相关基因胚系突变或体细胞突变以及表观遗传失活等诸多因素导致。当HRD存在时, DNA双链断裂会过度依赖非同源末端连接、微同源末端连接和单链退火途径等低保真、高易错的替代性DNA损伤修复途径, 从而极可能造成核酸序列的插入/缺失, 拷贝数异常, 并引起染色体交联, 造成基因组和染色体不稳定。
HRD会产生特定的、可量化的、稳定的基因组改变, 可通过建立基于基因组特征分析的评估体系来预测肿瘤HRD状态及其程度, 已成为晚期卵巢癌患者临床应用PARP抑制剂的新型生物标志物, 也可能对乳腺癌、前列腺癌等肿瘤的PARP抑制剂和铂类药物的临床用药具有指导价值。
HRD检测采用NGS方法, 通常包括两个部分, BRCA1/2突变状态及基因组不稳定性状态的评分(genomic instability score, GIS), 或称HRD评分(HRD score)。
对于后者, 一般通过对细胞内SNP进行检测和计算得出。其原理是基于细胞内因HRD而引起的DNA损伤, 将以一些特定且可识别的方式在基因组上留下痕迹, 如LOH、端粒等位基因失平衡(telomeric allelic imbalance, TAI)和大片段迁移(large-scale state transitions, LST)。
截至目前, 全球范围内仅2款HRD检测产品在大型Ⅲ 期临床研究中得到验证:Myriad myChoice® CDx和FoundationFocusTM CDx BRCA LOH。2019年10月, Myriad myChoice® CDx首次被FDA批准作为鉴别HRD阳性的晚期上皮性卵巢癌的伴随诊断; 2020年5月, Myriad myChoice® CDx被FDA批准作为鉴别HRD阳性的晚期卵巢癌患者奥拉帕利一线维持治疗的伴随诊断, 用于筛选奥拉帕利联合贝伐单抗治疗的潜在临床获益人群。
而FoundationFocusTM CDx BRCA LOH尚未获批伴随诊断。
2020年8月, Myriad myChoice® CDx被ASCO在《PARP inhibitors in the management of ovarian cancer:ASCO guideline》中推荐, 也是唯一提名的商业伴随诊断产品。FDA获批的Myriad myChoice® CDx通过BRCA1/2的致病性变异状态及GIS来评价HRD状态。目前关于Myriad myChoice® CDx的报告解读已有较明确的标准:当BRCA1/2突变和(或)GIS评分≥ 42分时, 即判定为HRD阳性, 详见表4。
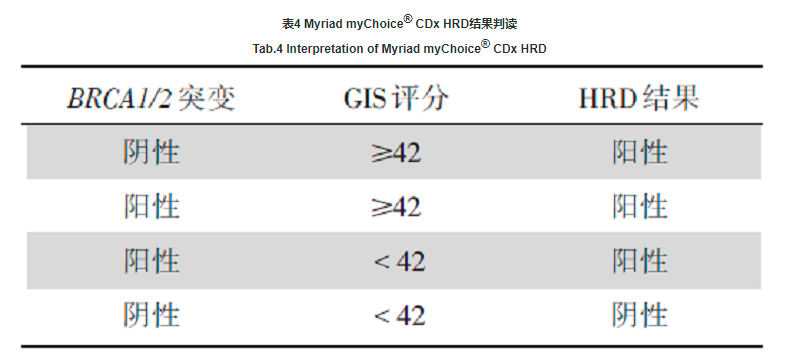
最终基于患者HRD状态, 国内外指南及共识的推荐、PARP抑制剂国内外药物适应证, 指导临床合理用药。
资料来源:二代测序临床报告解读肿瘤学专家组. 肿瘤二代测序临床报告解读共识. 循证医学, 2022,22(2): 65-79.
内容来源:基因谷