
全球找新药”——为生命寻找新希望的大型实时在线招募平台

出国抗癌就医,找全球肿瘤医生 为家人寻找美国和日本治疗方案

癌症疫苗突破 横扫九大癌种

药企入驻新药小程序啦
方舟新药-帮助患者寻找新药治疗的希望

肿瘤患者免疫 细胞功能自我监测

专业抗癌就医服务机构




擅长:黑色素瘤、免疫治疗、靶向治疗

哈佛大学附属麻省总医院
擅长:卵巢癌、宫颈癌、子宫癌,新型靶向药物和个体化治疗

Winthrop P. Rockefeller 癌症研究所
擅长:宫颈癌、卵巢癌 、子宫内膜癌、子宫癌

哈佛医学院
擅长:肉瘤、胃肠道间质瘤、脊索瘤和硬纤维瘤患者的医疗管理

丹娜法伯癌症研究所
擅长:骨肉瘤 胃肠道间质瘤 (GIST) 软组织肉瘤

斯坦福大学肿瘤研究所
擅长:乳腺癌、分子疗法、内科肿瘤学

麻省总医院
擅长:结直肠癌、胰腺癌、胃癌、食管癌、胆囊癌、肝癌

美国德克萨斯大学安德森癌症中心
擅长:

《CAR-T三人行》|第十五期:2025年4月24日16点,魏双教授:2025肺癌治疗开启新篇章!CAR-T等创新疗法带来新机"愈"



胃癌治疗新技术,电场疗法联合化疗一线治疗胃癌新数据公布,客观缓解率50%




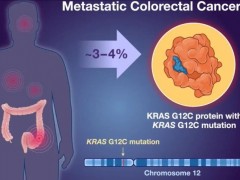
KRAS阳性肠癌新药AMG510(Lumakras、Sotorasib)即将于2025年初上市,30%患者肿瘤靶病灶显著缩小


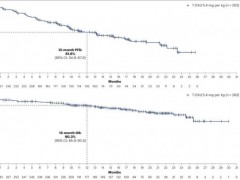
靶向HER2的抗体偶联(ADC)药物德曲妥珠单抗帮助乳腺癌脑转移的患者无进展生存率超60%



癌愈未来--全国肿瘤防治宣传周第九期:领衔突破!创新CAR-T疗法重塑复发、难治、罕见淋巴瘤治疗格局!
13次播放

癌愈未来--全国肿瘤防治宣传周第七期:CAR-T利器—为自身免疫疾病患者带来新希望
25次播放

癌愈未来--全国肿瘤防治宣传周第六期:CAR-T治疗复发难治淋巴瘤新进展
28次播放

癌愈未来--全国肿瘤防治宣传周第五期:2025抗癌新势力——TIL疗法带来生存突破
35次播放

癌愈未来--全国肿瘤防治宣传周第四期:免疫干预在肿瘤综合治疗中的重要地位
40次播放

癌愈未来--全国肿瘤防治宣传周第三期:实体肿瘤细胞免疫治疗前沿
29次播放

《CAR-T三人行》第十四期:中国CAR-T领跑全球:2025强势突围胃癌、胰腺癌等实体肿瘤!
37次播放

癌愈未来--全国肿瘤防治宣传周第一期:癌症筛查怎么做才有效?告诉14筛查和诊治亿中国人如何科学防癌
34次播放




益康素短肽全营养素

维生素C粉

胸腺蛋白肽粉

雪舞白舞茸,含舞茸多糖

美源康富ABD活性因子























































































 首页
首页 咨询
咨询 方舟新药
方舟新药 营养商城
营养商城