


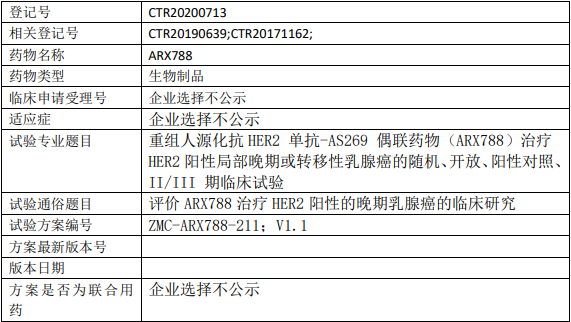
浙江医药ARX788临床试验,评价重组人源化抗HER2单抗-AS269抗体偶联药物(ARX788)治疗HER2阳性的晚期乳腺癌临床试验
1、试验目的
评估ARX788相较于拉帕替尼联合卡培他滨治疗HER2阳性局部晚期或转移性乳腺癌的有效性。
2、试验设计
试验分类:安全性和有效性
试验分期:其它
设计类型:平行分组
随机化:随机化
盲法:开放
试验范围:国内试验
3、受试者信息
年龄:18 岁(最小年龄)至 75 岁(最大年龄)
性别:男+女
健康受试者:无
入选标准
1.受试者或其法定代理人自愿书面签署知情同意书;2.年龄为 18~75 周岁(包含上下限),男性或女性;
3.经细胞学或组织学诊断为乳腺癌,且为不可切除的局部晚期或转移性疾病者;
4.既往针对复发或转移性疾病接受过≤2 线化疗(不包括激素治疗)的受试者;
5.经研究者确认或病史记录具有明确的疾病进展,包括最近一次针对局部晚期或转移性疾病治疗期间或之后肿瘤疾病进展或在乳腺癌(新)辅助治疗期间或治疗结束 12 个月内疾病进展者;
6.既往辅助治疗、局部晚期或转移性疾病曾接受过至少一次曲妥珠单抗治疗;
7.既往接受过紫杉类药物治疗;
8.根据 RECIST1.1 标准,至少有一个可测量的靶病灶;
9.筛选期内可提供经中心实验室认定合格的用于 HER2 检测的组织样本;
10.组织样本经中心实验室检测确定为 HER2 阳性(定义为 ICH3+或 FISH 基因扩增比率≥2.0);
11.ECOG 功能状态评分 0~1 分;
12.筛选期超声心动图检测 LVEF≥50%;
13.已从既往外科手术和既往癌症治疗相关的任何 AE 中恢复,以下情况除外:
a. 脱发;
b. 色素沉着;
c. 放疗引起的远期毒性,经研究者判断不能恢复;
d. 铂类引起的 2 级及以下神经毒性;
14.足够的骨髓、肝、肾及凝血功能(可参考各临床试验中心正常值上限):
骨髓(筛选检查前 2 周内未输血或使用辅助白细胞、血小板增加药物):绝对中性粒细胞计数(ANC)≥1.5×10^9/L;血小板≥100×10^9/L;血红蛋白≥90g/L;
肝功能:血清总胆红素≤1.5 倍正常值上限(ULN);丙氨酸氨基转移酶(ALT)、天门冬氨酸氨基转移酶(AST)≤3 倍 ULN,碱性磷酸酶(ALP)≤2.5倍 ULN;有肝转移时 ALT、AST≤5 倍 ULN,有骨转移时,ALP≤5倍 ULN;
肾功能:血清肌酐≤1.5 倍 ULN;
凝血功能:国际标准化比率(INR)≤1.5 倍 ULN,活化部分凝血活酶时间(APTT)≤1.5倍 ULN;
15.预期生存期≥3 个月。
排除标准
1.已知对 ARX788 的任一活性成分或辅料过敏者,或有明确蛋白类药物过敏史、特异性变态反应病史(哮喘、风湿、湿疹性皮炎)或曾发生过其它严重的过敏反应;
2. 已知对卡培他滨和拉帕替尼的某些成分或类似药物有超敏反应或迟发型过敏反应,或已知有卡培他滨使用禁忌症者,主要包括既往对氟嘧啶有严重、非预期的反应或已知对氟尿嘧啶过敏者和已知二氢嘧啶脱氢酶(DPD)活性完全缺乏者;
3. 试验用药品首次使用前 6 个月内使用过卡培他滨,或针对复发转移性乳腺癌使用含卡培他滨(包括其他氟尿嘧啶类药物)或拉帕替尼片方案在治疗过程中进展或治疗结束后 6 月内进展;针对辅助治疗中,含卡培他滨(包括其他氟尿嘧啶类药物)或拉帕替尼片方案结束 2 年内进展者;
4. 既往接受过 T-DM1 或其他 HER2-ADC 类药物治疗者;
5. 在过去 5 年内患有另一种恶性肿瘤者,接受治愈性治疗的原位宫颈癌、非黑色素瘤皮肤癌等恶性肿瘤除外;
6. 存在原发性中枢神经系统(CNS)恶性肿瘤或经局部治疗失败的CNS 转移患者,无症状脑转移、或临床症状稳定且首次给予试验用药品前无须类固醇激素和其他针对脑转移治疗≥28天的患者可以入组;
7. 存在活动性肺炎或间质性肺炎史或药物性肺部疾病者,放疗后3个月且无临床症状的放射性肺炎患者可以入组;
8. 现患角膜炎、角膜疾病、视网膜疾病或活动性眼部感染等需要干预的眼部疾病者;
9. 不愿或不能在试验期间停止佩戴角膜接触镜者;
10. 心功能不全受试者,包括但不限于充血性心力衰竭、透壁性心肌梗死受试者、需要药物治疗的心绞痛、临床上显著的心脏瓣膜病及高危心律失常或筛选期ECG检查中QTc异常且有临床意义(静息状态下, ECG 检 查 校 正 后 QTc > 450msec[ 男 ] 或 QTc>470msec[女]);
11. 未控制的高血压(收缩压>160mmHg 或舒张压>100mmHg);
12. 根据研究者判断,存在严重或不能控制的全身性疾病(如不稳定或不能代偿的呼吸、心脏、肝或肾脏疾病)证据的受试者;
13. 首次给予试验用药品前 4 周内使用过化疗、放疗、免疫治疗者(生理替代剂量糖皮质激素[泼尼松或等效物<15mg/天]允许使用);
14. 首次给予试验用药品前 2 周内使用过乳腺癌内分泌治疗者;
15. 首次给予试验用药品前 2 周内接受过针对骨转移的姑息性放疗者;
16. 既往暴露于蒽环类药物的累积达到以下剂量:多柔比星或脂质体多柔比星>500mg/m2;表柔比星>900mg/m2;米托蒽醌>120mg/m2;其他(即脂质体多柔比星或其他蒽环类药物>相当于 500mg/m2 的阿霉素);如果使用多于一种蒽环类药物,那么累积剂量不得超过相当于 500mg/m2 的阿霉素。
17. 任何不受控制的感染,或者其他可能限制试验依从性或干扰评价的情况;
18. 乙肝表面抗原阳性且 HBV DNA≥ULN 者,或丙型肝炎病毒抗体、梅毒螺旋体抗体、人类免疫缺陷病毒抗体检查结果中任意一种呈阳性者;
19. 首次使用试验用药品前 2 周内或试验期间计划接受重大外科治疗,或发生严重创伤性损伤者;
20. 妊娠期或哺乳期女性受试者;
21. 不愿或不能在本试验的整个治疗期间及试验用药品末次给药后 8 个月内采用可接受的方法进行避孕的育龄妇女(育龄妇女包括:任何有过月经初潮,以及未接受过成功的人工绝育手术[子宫切除术、双侧输卵管结扎、或双侧卵巢切除术]或未绝经)或男性受试者;
22. 首次给予试验用药品前 4 周内参与临床试验并使用了其他试验药物;
23. 任何精神或认知障碍,可能会限制其对知情同意书的理解、执行;
24. 研究者认为不适合参与本试验的其他情况,如依从性差等。
研究者信息
1、主要研究者信息
姓名:胡夕春
学位:医学博士
职称:主任医师
单位名称:复旦大学附属肿瘤医院
2、各参加机构信息
机构名称:复旦大学附属肿瘤医院
(主要)研究者:胡夕春
国家:中国
省(州)-城市:上海-上海






















 首页
首页 咨询
咨询 方舟新药
方舟新药 营养商城
营养商城