


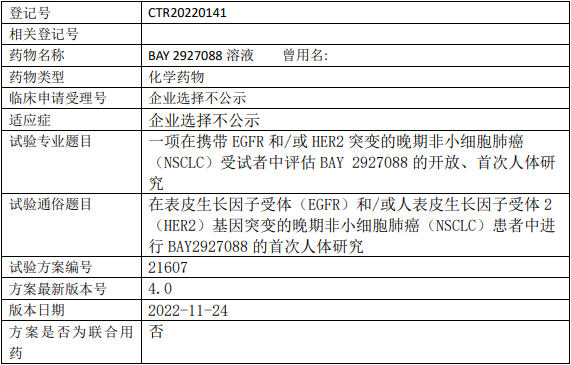
拜耳医药BAY2927088临床试验,评估BAY 2927088溶液治疗携带EGFR和/或HER2突变的晚期非小细胞肺癌的I期临床试验
试验目的
主要目的
1、确定BAY 2927088口服给药的安全性和耐受性;
2、确定BAY 2927088的最大耐受剂量(MTD)或最大给药剂量(MAD);
3、表征BAY 2927088的药代动力学(PK)。
次要目的
1、评估BAY 2927088的初步抗肿瘤活性;
2、确定BAY 2927088的II 期推荐剂量(RP2D)。
其他目的
1、评价BAY 2927088的抗肿瘤活性;
2、评价生物标志物及其临床结局的关系;
3、考察PK/暴露与生物标志物之间的关系;
4、考察PK/暴露与临床结局(例如:安全性/耐受性、ECG 和有效性)之间的关系;
5、评估BAY 2927088液体溶液制剂(LSF)和片剂的相对生物利用度。
试验设计
试验分类:药代动力学/药效动力学试验
试验分期:I期
设计类型:单臂试验
随机化:非随机化
盲法:开放
试验范围:国际多中心试验
受试者信息
年龄:18岁(最小年龄)至无上限(最大年龄)
性别:男+女
健康受试者:无
出入排标准
入选标准
1、筛选时经组织学或细胞学证实的局部晚期NSCLC(不适合接受决定性治疗)或者复发/转移性NSCLC(不包括小细胞型或混合组织学)。
2、已证实既往至少接受过一种全身疗法治疗晚期疾病后出现疾病进展。因任何原因无法使用标准治疗、无法耐受标准治疗、不适合接受标准治疗的受试者也可能适合入组本研究。
3、足够的存档肿瘤组织(最好在末次靶向治疗后采集,且采集时间不超过6个月),原发部位或转移部位均可。如果无可用存档材料,应在可行且该程序不会对受试者造成重大风险的情况下进行新鲜肿瘤活检。
4、符合RECIST v1.1标准的可测量病灶,在筛选期间(如果筛查期间进行了活检)至少有一个病灶没有被选择进行活检,可以在基线时通过计算机断层扫描(CT)或磁共振成像(MRI)准确测量且适合准确重复测量的病灶。活检病灶不应作为RECIST 1.1肿瘤评估的靶病灶。既往接受过放疗的病灶必须显示进展才能认为可测量。
5、经临床实验室改进修正案(CLIA)认证(美国[US]研究中心)或同等认证的(美国以外地区)当地实验室评估,证实携带激活的EGFR和/或HER2突变。
6、美国东部肿瘤协作组(ECOG)体能状态评分为0或1
7、预期寿命至少12周
8、在研究治疗首次给药前7天内通过以下实验室检查评估具有足够的骨髓功能:
a. 输注血红蛋白≥9.0 g/dL。符合标准,无促红细胞生成素依赖性,而且在检测前2周内未输注浓缩红细胞(pRBC)。
b. 血小板≥100×109细胞/L。
c. 中性粒细胞绝对计数≥1.5×109个细胞/L。符合标准且在检测前2周内未使用造血生长因子(例如G-CSF)。
9、研究治疗首次给药前7天内通过以下实验室检查评估认为具有足够的肾功能:根据肾脏疾病饮食改良研究组(MDRD)公式,估计肾小球滤过率(eGFR)>60 mL/min/1.73 m2
10、研究治疗首次给药前7天内通过以下实验室检查评估认为具有足够的肝功能:
a. 总胆红素≤1.5×ULN(或对于确诊Gilbert-Meulengracht综合征的受试者或认为高胆红素血症是由肝转移引起的受试者,则≤3×ULN);
b. 天冬氨酸氨基转移酶和丙氨酸氨基转移酶≤2.5×ULN(或如果是由肿瘤累及肝脏造成,则≤5×ULN)。
排除标准
1、研究药物首次给药前≤8天或终末期消除半衰期的5倍(以较短者为准)接受过EGFR酪氨酸激酶抑制剂(TKI)治疗。
2、研究药物首次给药前≤14天内接受过全身抗癌治疗(不包括上述EGFR TKI)。
3、研究药物首次给药前,放疗、立体定向放射治疗(SRS)和姑息放疗≤14天。
4、在研究药物首次给药前≤28天接受过免疫治疗。
5、既往抗癌治疗后出现的任何≥2级毒性尚未缓解,脱发和皮肤色素沉着除外。在研究者与和申办方协商一致后,有稳定的慢性2级毒性的受试者可以入组。
6、有任何原发性脑或柔脑膜疾病(症状性或无症状性)病史,存在症状性中枢神经系统(CNS)转移,或需要局部治疗(如放疗或手术)的CNS转移。
7、有脊髓压迫或脑转移病史,但以下情况除外:
a. 筛选时无症状且在BAY 2927088首次给药前至少7天停用或接受低剂量皮质类固醇治疗(≤10 mg泼尼松或等效药物)的脑转移受试者有资格入组剂量递增和回填阶段;
b. 筛选时无症状的脑转移经治受试者如果符合以下所有标准,则有资格进入剂量扩展阶段: 筛选期间通过临床检查和脑成像(MRI或CT)确定,CNS靶向治疗后至少4周内无进展证据(新发脑转移瘤或脑转移瘤增大)。受试者必须在BAY 2927088首次给药前7天停用或接受低剂量皮质类固醇(≤10 mg泼尼或等效药物;
c. 在确定性治疗后有>3个月脊髓压迫病史且在筛选期间影像学(MRI或CT)显示疾病稳定且临床无症状的受试者。
8、有纽约心脏病协会(NYHA)心功能分级>II级充血性心力衰竭(CHF)史或有需要治疗的严重心律失常(如室性心律失常、房颤)或存在任何具有临床重要的心律、传导或形态或静息ECG异常(例如:完全左束支阻滞、三度心传导阻滞、二度心传导阻滞、PR间期>250 msec)。
9、受试者患有以下疾病:
a. 已知感染人类免疫缺陷病毒(HIV),以下情况除外:如果符合以下条件,有HIV感染史的受试者有资格参加研究,具体由研究者酌情决定:CD4+T细胞(CD4+)计数≥350个细胞/uL。受试者在开始研究药物治疗前已接受至少4周的既定抗逆转录病毒治疗(ART),而且在开始研究治疗前的HIV病毒载量低于400拷贝/mL。 正在使用的ART不含强效CYP3A4诱导剂或抑制剂,预计不会与研究药物产生重叠毒性。受试者在过去12个月内未发生机会性感染;
b. 活动性乙型肝炎感染(乙型肝炎表面抗原[HbsAg]和乙型肝炎病毒[HBV]DNA阳性);
c. 活动性丙型肝炎感染(抗HCV抗体阳性和定量HCV RNA结果大于试验的检测下限);
10、研究药物首次给药前14天内使用强效CYP3A4抑制剂和诱导剂。本研究期间禁止使用强效CYP3A4抑制剂和诱导剂,直至安全性随访访视。






















 首页
首页 咨询
咨询 方舟新药
方舟新药 营养商城
营养商城