时间:2021-05-20 11:59 编辑:全球肿瘤医生网
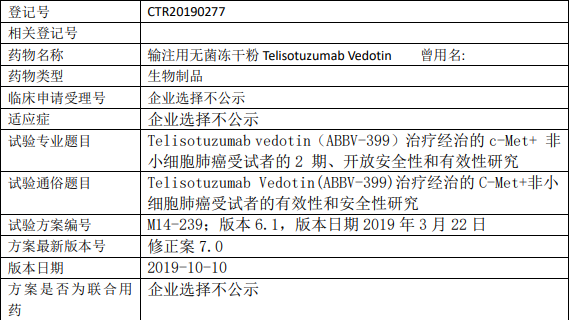
Telisotuzumab Vedotin(ABBV-399)临床试验,Telisotuzumab Vedotin(ABBV-399)治疗经治的C-Met+非小细胞肺癌受试者的有效性和安全性研究
1、试验目的
主要目的:-本项研究的主要目的是确定 telisotuzumab vedotin 在 c-Met+ NSCLC 受试者中的总缓解率(ORR)。
次要目的: -缓解持续时间(DoR) -疾病控制率(DCR) -疾病控制持续时间(DDC) -无疾病进展生存期(PFS) -总生存期(OS) -安全性和耐受性
2、试验设计
试验分类:安全性和有效性
试验分期:II 期
设计类型:单臂试验
随机化:非随机化
盲法:开放
试验范围:国际多中心试验
3、受试者信息
年龄:18 岁(最小年龄)至不适用岁(最大年龄)
性别:男+女
健康受试者:无
入选标准
1.在开始任何筛选步骤或研究特定步骤之前,受试者或其合法代理人必须自愿签署由机构审查委员会(IRB)/机构审查委员会(IEC)批准的知情同意书。在确认为当地要求的情况下时,受试者(非代理人)必须提供书面同意。
2.成年男性或女性,18 岁或以上。
3.受试者必须存在 c-Met+ NSCLC,由艾伯维指定的 IHC 实验室评估,或是存在已知的文件证实的 MET 基因扩增,其定义为: ?MET/CEP7 比值2(荧光原位杂交(FISH)检测法,基于至少 50个肿瘤细胞的平均值);或是 FoundationOne检测肿瘤组织或血浆中的循环肿瘤 DNA[ctDNA]、Guardant360或PDGx检测血浆中的 ctDNA 有明确扩增结果。 注:如果 MET基因扩增是通过任何其他方法或 FISH 标准测定的,则必须提供文档并由艾伯维 TA MD 审查。
4.不存在文件证实的MET 基因扩增的受试者必须在预筛选期间提供用于艾伯维指定的 IHC 实验室评估 c-Met 水平的存档或者新鲜的肿瘤组织。如果受试者的 c-Met 蛋白表达水平符合入选标准是基于存档组织的检测结果,则受试者必须同意在入组之前提供新鲜肿瘤组织,以评估 c-Met 蛋白表达水平。
5.存在已知的文件证实的 MET 基因扩增的受试者可以进行预筛选。入组前将需要采集用于艾伯维指定的 IHC 实验室评估 c-Met蛋白表达水平的存档或者新鲜的可评估肿瘤组织(在测定 MET 扩增状态的同时或之后获得)。
6.受试者具有足够的骨髓、肾脏和肝脏功能,如下: ?骨髓:中性粒细胞绝对计数(ANC) 1,000/mm3,血小板100,000/mm3;血红蛋白9.0 g/dL; ? 肾功能:血清肌酐1.5 机构的正常值上限(ULN),或者采用 24 小时尿液测定或采用 Cockcroft-Gault 公式计算的肌酐清除率 50 mL/分钟:CrCl(mL/分钟) = (140 年龄) (体重,kg)( 0.85,如果为女性)72 血清肌酐(mg/dL)?肝功能:胆红素 1.5 ULN,天冬氨酸氨基转移酶(AST)、 且丙氨酸氨基转移酶(ALT) 3.0 ULN,-谷氨酰转移酶(GGT) 5 ULN、且白蛋白 3.0 g/dL。 ? 存在肝转移的受试者的肝功能:胆红素 1.5 ULN,AST、ALT 和 GGT 5 ULN、且白蛋白 3.0 g/dL。
7.受试者愿意且能够依从本研究方案中要求的研究步骤。
8.受试者具有组织学证实的、EGFR 状态明确的(野生型或突变型;含位点状态明确)非鳞状细胞 NSCLC;或者具有组织学证实的鳞状细胞 NSCLC。值得注意的是,存在其他可诉性突变的受试者,只要 EGFR 状态已知且符合所有其他入选标准,也可以入组。
9.受试者必须为局部晚期或转移性 NSCLC。
10.受试者的美国东部肿瘤协作组(ECOG)体能状态为 0 或 1。
11.根据第 1.1 版 RECIST,受试者必须存在可测量病灶。
12.受试者不得有腺鳞癌病史。
13.局部晚期或转移性 NSCLC 受试者不得接受超过二线的既往系统化疗(包括不得接受超过一线的系统性细胞毒化疗)。 ? 就本入选标准来说,作用于相同 TK 靶点的多线 TKIs 按一线治疗计。
14.受试者经过系统性细胞毒化疗(或不适合系统性细胞毒化疗)和免疫检查点抑制剂(如单药治疗或联合全身性细胞毒化疗,或不适合免疫检查点抑制剂),以及既往针对驱动基因改变的抗癌疗法(如适用)后疾病仍然进展。
15.受试者不应接受过既往 c-Met 靶向抗体疗法。
16.存在中枢神经系统(CNS)转移灶的受试者仅在接受针对性治疗(例如手术或放疗)之后且符合以下条件才可以入组: ? 接受针对性治疗之后至少 4 周未见 CNS 转移灶进展的证据。? 无症状并且在 telisotuzumab vedotin 首次给药前停用类固醇全身治疗或抗惊厥药物治疗至少 2 周。
17.受试者不得存在需要接受全身性类固醇治疗的间质性肺病或肺炎的病史,或有任何证据提示活动性间质性肺病或肺炎。
18.受试者不得存在既往抗癌治疗后尚未缓解的具有临床意义的 2 级不良事件,脱发或贫血除外。
19.受试者在 telisotuzumab vedotin 首次给药前 21 天内不得接受大型手术治疗。
20.受试者不得存在具有临床意义的疾病,包括但不限于以下:? 2 级水肿和淋巴水肿。 ? 2 级腹水和胸腔积液。 ? 2 级周围神经病变或 3 级周围神经病变病史。 ?尚未得到控制的活动性细菌或病毒感染 ? 纽约心脏病协会 III 级充血性心力衰竭 ?不稳定型心绞痛或心律失常
21.受试者不得存在可能影响研究依从性的精神疾病/社会问题。
22.受试者不得存在对任何含IgG 药物具有重大免疫反应的病史。
23.受试者不得存在研究者或者治疗领域医学总监(TA MD)认为可能会给受试者带来无法接受的较高毒性作用发生风险的任何医学疾病。
24.对于所有具有生育能力的女性,筛选访视时的血清妊娠检测结果为阴性,研究药物首次给药前基线时的尿液妊娠检测结果为阴性。
25.具有生育能力的女性受试者,从研究第 1 天至研究药物末次给药后至少 6 个月必须至少采用 1 种研究方案规定的避孕方法。
26.女性受试者没有正在妊娠、哺乳或者在研究期间或研究药物末次给药后约 6 个月内考虑妊娠。
27.与具有生育能力的女性配偶性生活活跃的男性,必须同意从研究第 1 天至研究药物末次给药后 6 个月内采取研究方案规定的避孕措施。
28.男性受试者没有考虑在研究期间或研究药物末次给药后约 6 个月内授孕或捐精。29.受试者不得在研究药物首次给药前 30 天内接受过任何活疫苗。
30.在 telisotuzumab vedotin 首次给药之前,在以下标注时间段内接受以下任何疗法治疗的,予以排除:
1周(7 天)内:接受草药或强效细胞色素 P450 3A4(CYP3A4)抑制剂治疗。
2周(14 天)内:半衰期7 天的小分子靶向药物;不涉及胸腔的放疗。
4周(28 天)内:全身细胞毒性化疗;半衰期7 天的小分子靶向药物;单克隆抗体、抗体-药物偶联物、放射性免疫偶联物或者是基于 T 细胞或其它细胞的疗法;涉及胸腔的放疗。接受以下任一疗法治疗不需要清洗期: ? 针对骨骼、皮肤或皮下转移灶的 10 处或以下姑息性放疗;中枢神经系统(CNS)转移灶见上。 ? 当前接受 EGFR TKIs 治疗的受试者。
排除标准
1.受试者必须符合以上所有入选标准,才能入组本项研究。如果受试者出现上述问题的否定回答,则将不能入组本项研究。
研究者信息
1、主要研究者信息
姓名:陆舜
学位:医学博士
职称:主任医师、教授
单位名称:上海市胸科医院
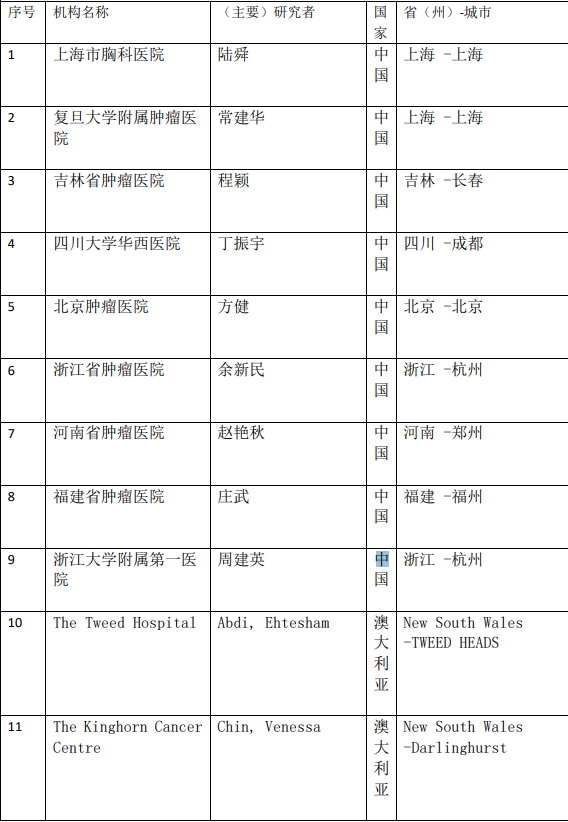
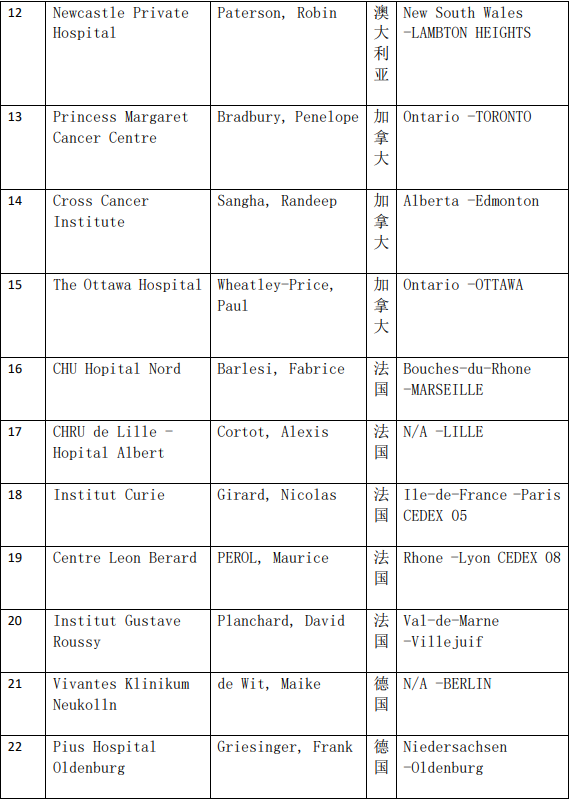
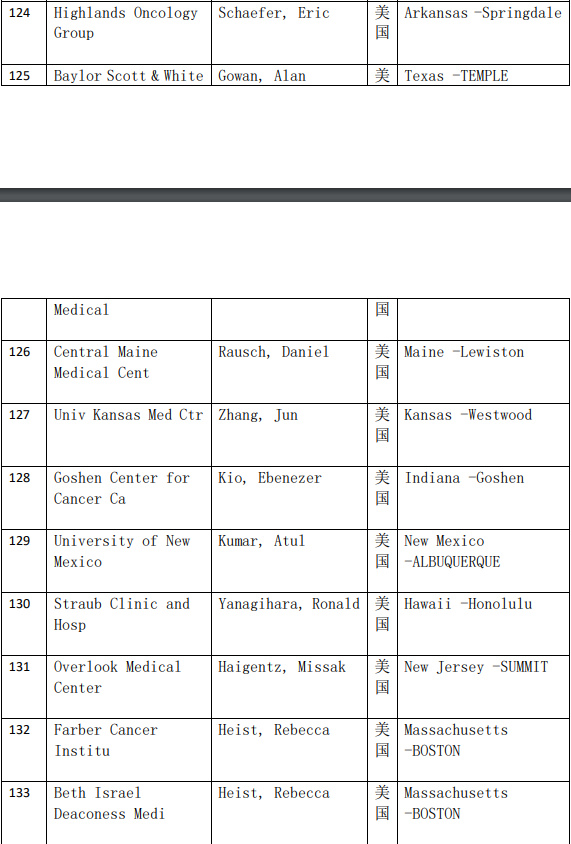
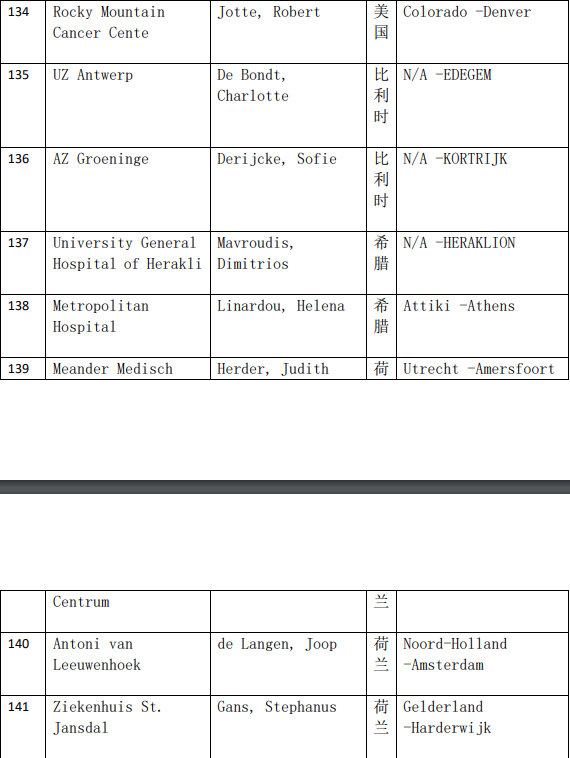
2、各参加机构信息
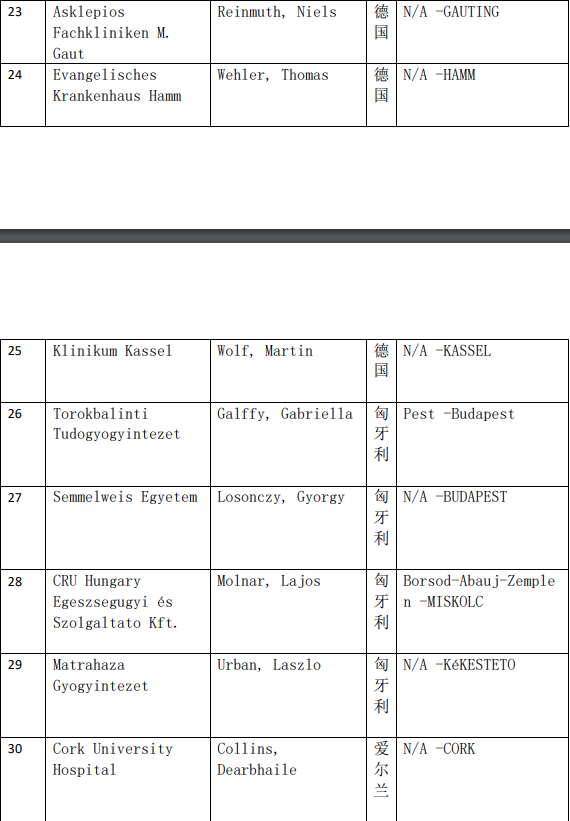
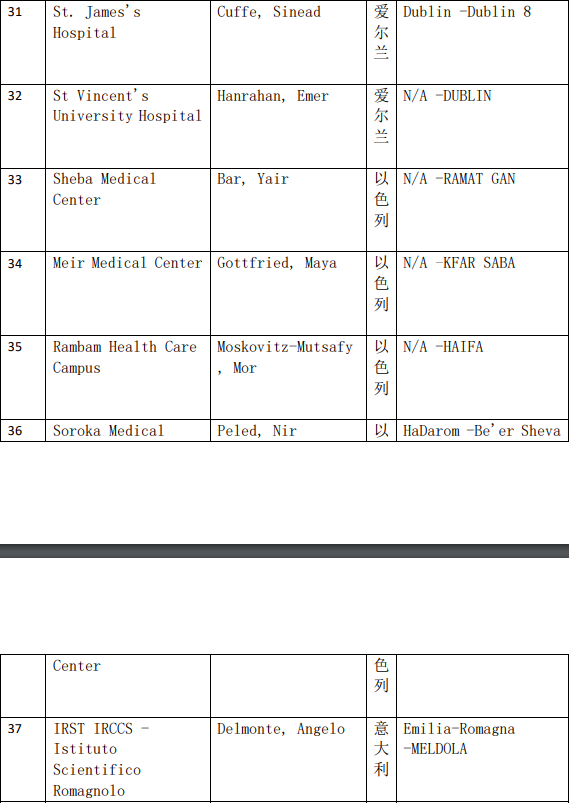
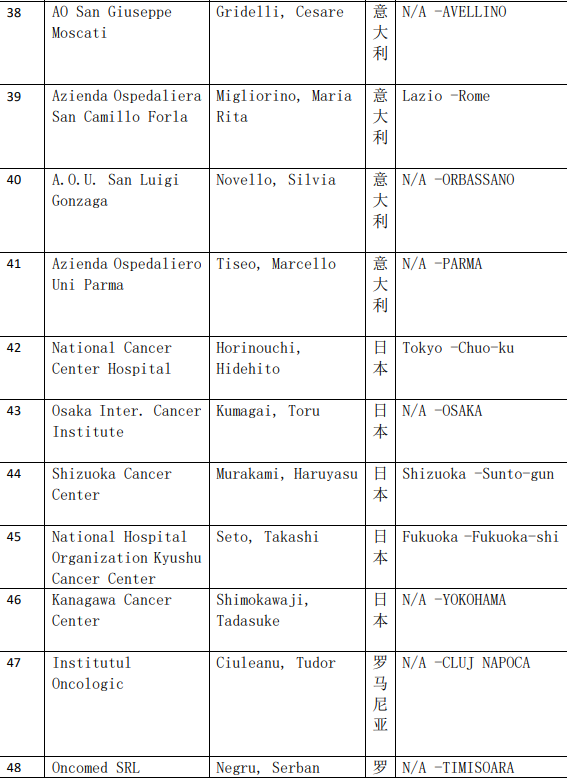
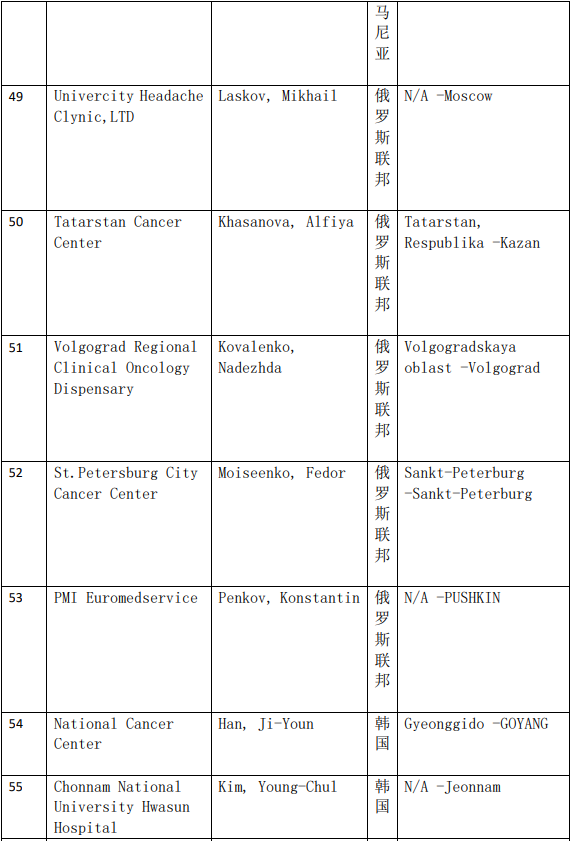
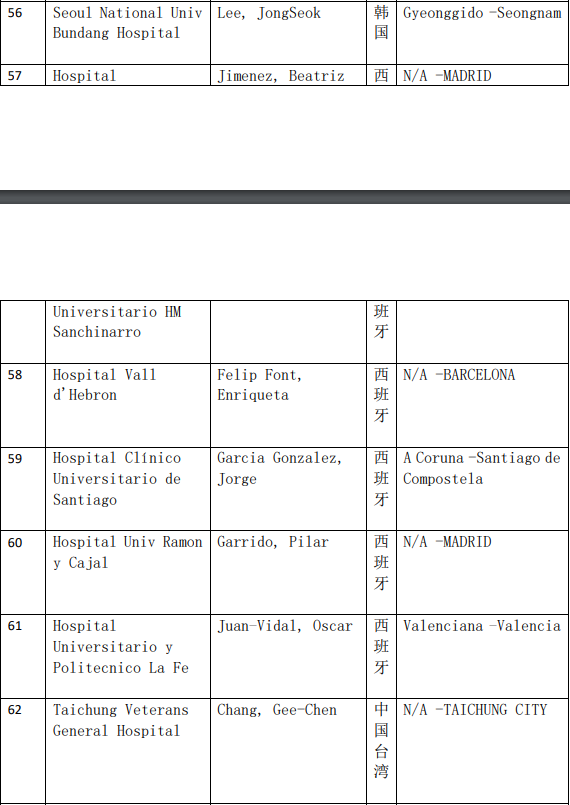
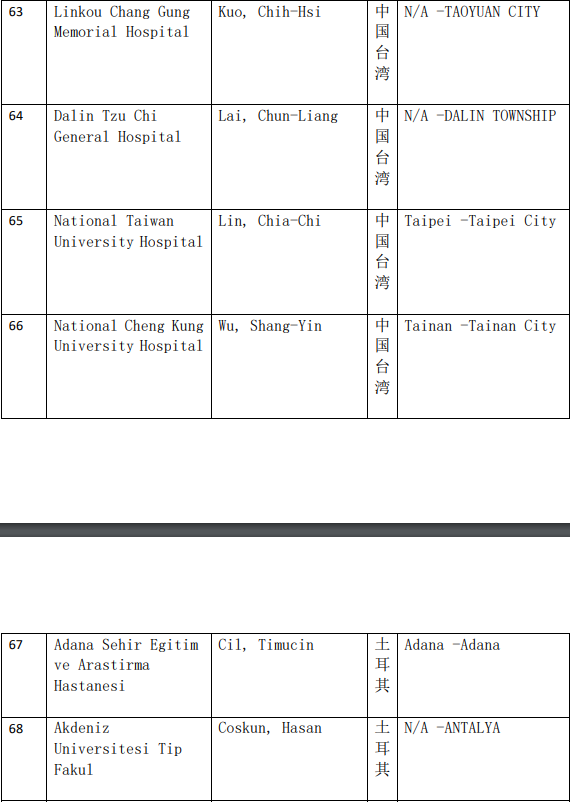
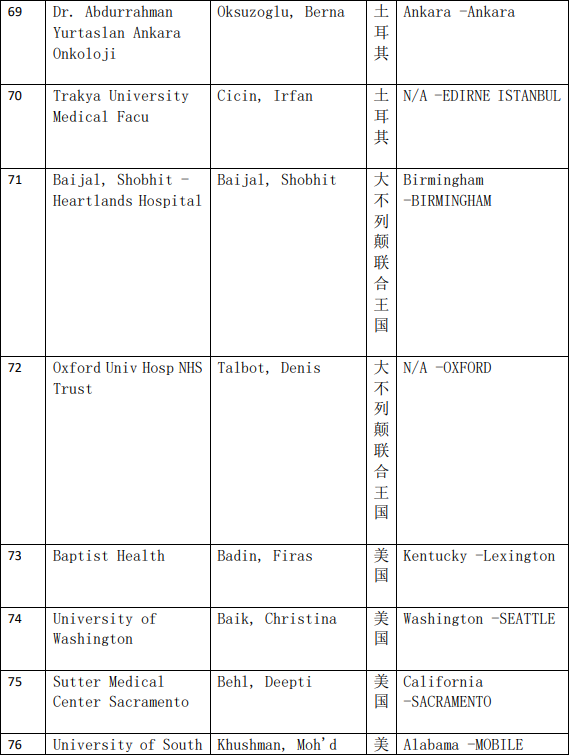
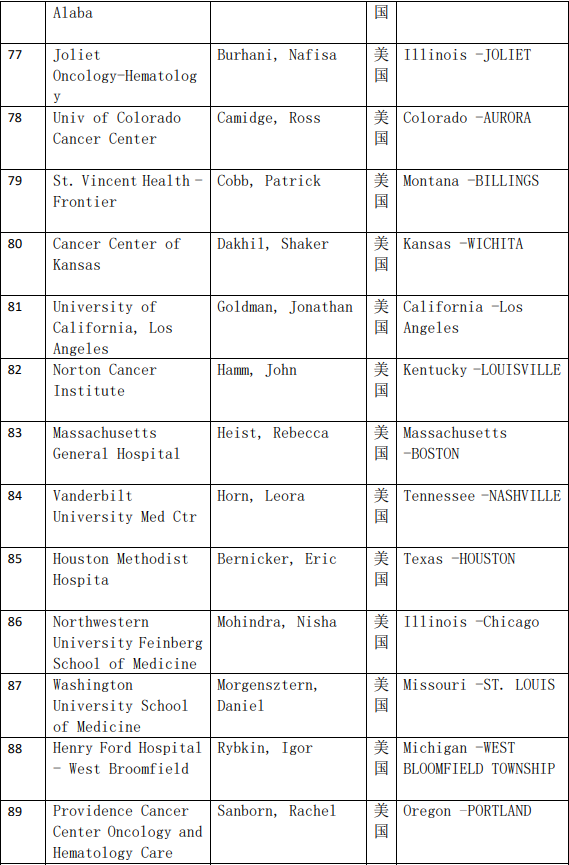
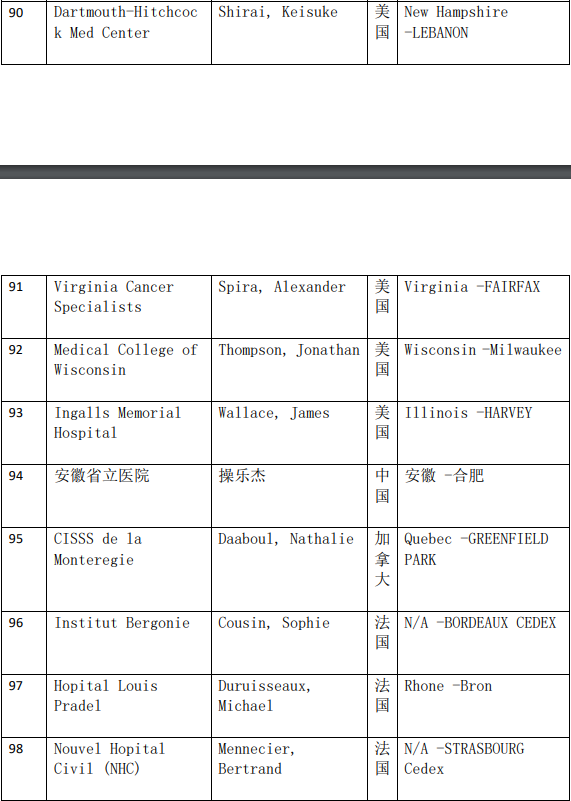
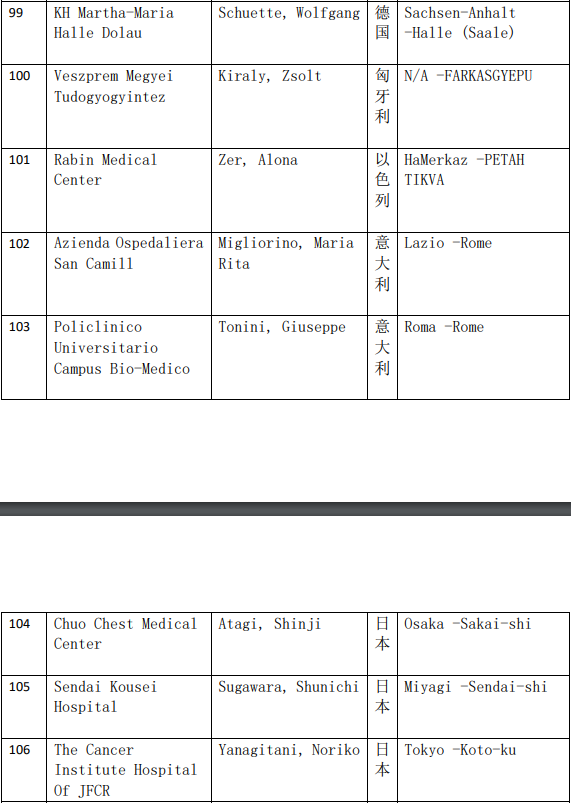
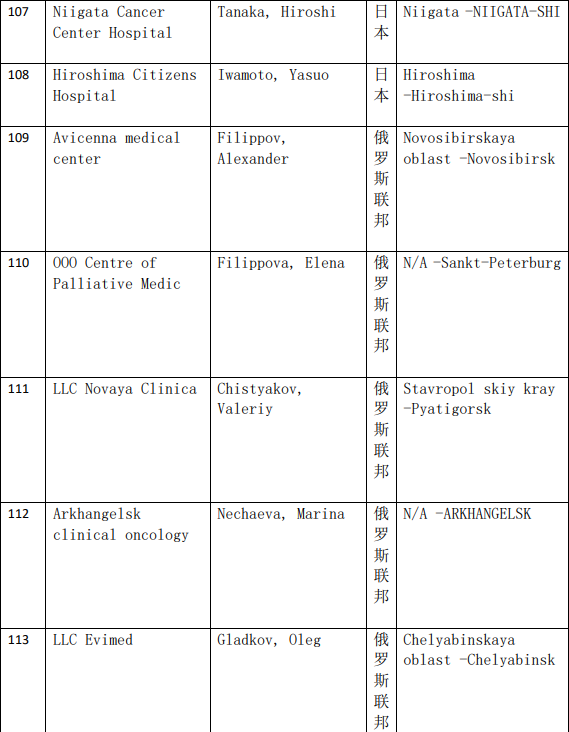
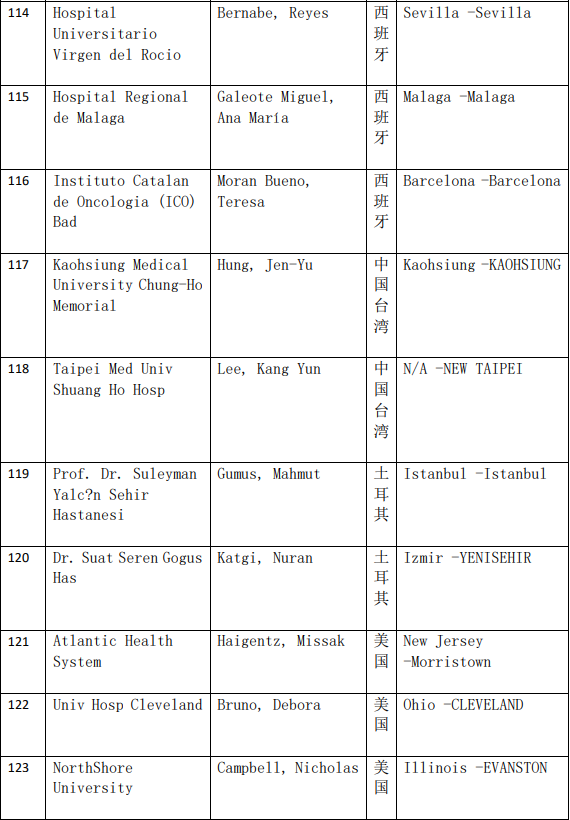
本网站新闻资讯、文章、研究数据、治疗案例均来自于国内外医学论文,所涉及到的新药、新技术有可能还处于临床研究阶段,患者不能作为治疗疾病的依据。癌症治疗目前尚无治愈手段,患者需要在医生的指导下,在医院接受正规治疗或参加新药新技术临床试验。









